-
Notifications
You must be signed in to change notification settings - Fork 0
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
Merge branch 'master' of https://github.molgen.mpg.de/heller/ssHMM
- Loading branch information
Showing
1 changed file
with
14 additions
and
199 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -1,214 +1,29 @@ | ||
| ## ssHMM - Sequence-structure hidden Markov model | ||
|
|
||
| A motif finder for sequence-structure binding preferences of RNA-binding proteins. | ||
| ssHMM is an RNA motif finder. It recovers sequence-structure motifs from RNA-binding protein data, such as CLIP-Seq data. | ||
|
|
||
| RNA-binding proteins (RBPs) play a vital role in the post-transcriptional control of RNAs. They are known to recognize RNA molecules by their nucleotide sequence as well as their three-dimensional structure. ssHMM is an RNA motif finder that combines a hidden Markov model (HMM) with Gibbs sampling to learn the joint sequence and structure binding preferences of RBPs from high-throughput RNA-binding experiments, such as CLIP-Seq. The model can be visualized as an intuitive graph illustrating the interplay between RNA sequence and structure. | ||
| ### Background | ||
|
|
||
| ### Overview | ||
| RNA-binding proteins (RBPs) play a vital role in the post-transcriptional control of RNAs. They are known to recognize RNA molecules by their nucleotide sequence as well as their three-dimensional structure. ssHMM combines a hidden Markov model (HMM) with Gibbs sampling to learn the joint sequence and structure binding preferences of RBPs from high-throughput RNA-binding experiments, such as CLIP-Seq. The model can be visualized as an intuitive graph illustrating the interplay between RNA sequence and structure. | ||
|
|
||
| ssHMM consists of 3 main scripts: | ||
| - **preprocess_dataset**: Prepares a CLIP-Seq dataset in BED format for ssHMM. It filters the BED file, fetches the genomic sequences, and predicts RNA secondary structures. | ||
| - **train_seqstructhmm**: Trains ssHMM on a given CLIP-Seq dataset and produces an intuitive visualization of the recovered motif. | ||
| - **batch_seqstructhmm**: Trains ssHMM on several different CLIP-Seq datasets and recovers one motif for each dataset. | ||
| ### Scope | ||
|
|
||
| ### Installation | ||
| ssHMM was developed for the analysis of data from RNA-binding assays. Its aim is to help biologists to derive a binding motif for one or a number of RNA-binding proteins. ssHMM was written in Python and is a pure command-line tool. | ||
|
|
||
| We distribute ssHMM both as a Python package and as a Docker image (similar to a virtual machine). The Docker image has already all dependencies installed and is much easier to use. When using the Python package, all dependencies have to be installed manually. | ||
| ### Output | ||
|
|
||
| #### Installation via Docker | ||
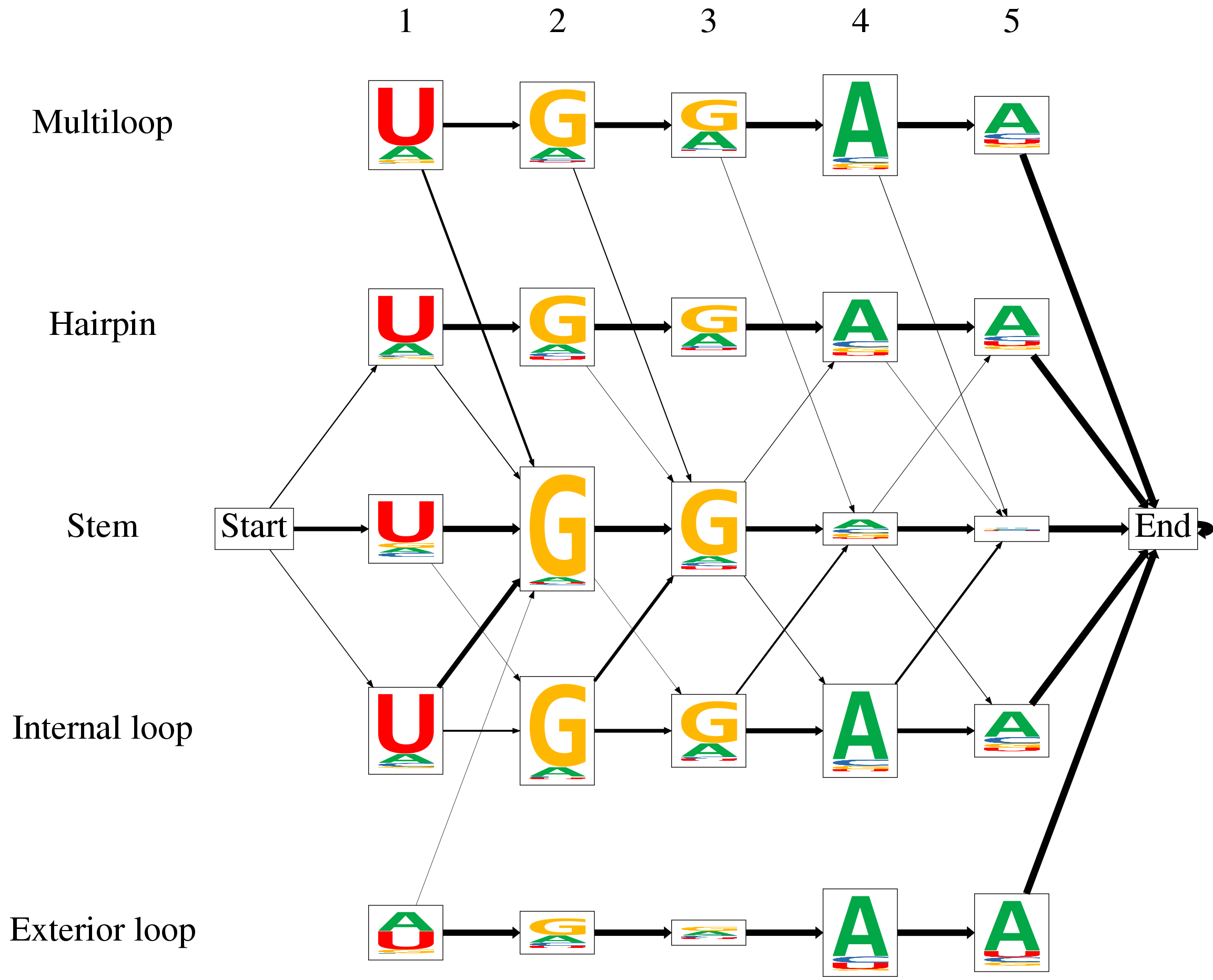
| Below, you find an example output of ssHMM. After training on an RBP dataset, the model can be visualized as a graph. Each of the HMM states, including the start and the end state, is represented by a box. The states are arranged into rows and columns where the columns represent the motif positions and the rows represent the five structural contexts of RNA. Each state's emission probabilities are visualized as a sequence logo. The relative heights of the nucleotides in a stack represent their emission probabilities. The total height of all four nucleotides is scaled according to the information content of the state's emission distribution. The transition probabilities between the states are visualized as arrows. The thicker an arrow between two states, the more likely is a transition between the two. To reduce clutter, no arrow is shown if the transition probability is less than 0.05. The RBP visualized below prefers the sequence motif UGGAA and primarily binds stem regions of RNA. | ||
|
|
||
| 1. Install Docker on your platform as described on https://docs.docker.com/engine/getstarted/ | ||
| 2. Run Docker and check whether everything is working: ```docker version``` | ||
| 3. Run ```docker run -t -i -v hellerd/sshmm```. This will download the Docker image containing ssHMM (if you have not yet downloaded it) and will run it. Once the image has been started, you can access it via a command line interface. You can now run ssHMM, e.g. by typing ```train_seqstructhmm --help```. You can exit the image with ```exit```. | ||
| 4. To access data located on your machine (e.g. in ```/home/someuser/data```) from inside the image, use the ```-v``` option: ```docker run -t -i -v /home/someuser/data:/data hellerd/sshmm```. This will make your available inside the image in the ```/data``` directory. You can now run ssHMM on this data, e.g. ```train_seqstructhmm /data/sequences.fasta /data/shapes.txt```. | ||
|  | ||
|
|
||
| #### Installation as a Python package | ||
| ### Documentation | ||
|
|
||
| ##### Prerequisites: | ||
| - GHMM (http://ghmm.org/) | ||
| - GraphViz (http://www.graphviz.org/) | ||
| Check out our documentation: http://sshmm.readthedocs.io | ||
|
|
||
| For preprocessing BED files with preprocess_dataset (tested only on Linux and macOS): | ||
| - bedtools (https://github.com/arq5x/bedtools2) | ||
| - awk | ||
| - RNAshapes (http://bibiserv.techfak.uni-bielefeld.de/rnashapes) | ||
| - RNAstructure (http://rna.urmc.rochester.edu/rnastructure.html) | ||
| ### Citation | ||
|
|
||
| ##### Installation of prerequisites: | ||
| Heller, D., Krestel, R., Ohler, U., Vingron, M., & Marsico, A. (2016). ssHMM: [Extracting intuitive sequence-structure motifs from high-throughput RNA-binding protein data](http://dx.doi.org/10.1101/076034). bioRxiv, 076034. | ||
|
|
||
| 1. Install prerequisites for GHMM as described on http://ghmm.sourceforge.net/installation.html. The commands for Ubuntu are: | ||
|
|
||
| ```bash | ||
| sudo apt-get update | ||
| sudo apt-get install build-essential automake autoconf libtool | ||
| sudo apt-get install python-dev | ||
| sudo apt-get install libxml++2.6-dev | ||
| sudo apt-get install swig | ||
| ``` | ||
| 2. Download and unpack GHMM from https://sourceforge.net/projects/ghmm/ | ||
| 3. Install GHMM as described on http://ghmm.sourceforge.net/installation.html. The commands for Ubuntu are: | ||
|
|
||
| ```bash | ||
| cd ghmm | ||
| sh autogen.sh | ||
| sudo ./configure | ||
| sudo make | ||
| sudo make install | ||
| sudo ldconfig | ||
| ``` | ||
| 4. Install GraphViz. On Ubuntu: | ||
|
|
||
| ```bash | ||
| sudo apt-get install graphviz | ||
| sudo apt-get install libgraphviz-dev | ||
| ``` | ||
| 5. Install pip if not already installed. On Ubuntu: | ||
|
|
||
| ```bash | ||
| sudo apt-get install python-pip | ||
| ``` | ||
| 6. Install PyGraphViz: | ||
|
|
||
| ```bash | ||
| sudo PKG_CONFIG_ALLOW_SYSTEM_LIBS=OHYESPLEASE pip install pygraphviz | ||
| ``` | ||
| 7. Install bedtools as described on http://bedtools.readthedocs.io/en/latest/content/installation.html | ||
| 8. Download and install RNAshapes as described on http://bibiserv.techfak.uni-bielefeld.de/rnashapes?id=rnashapes_view_download. | ||
| 9. Download and install RNAstructure from http://rna.urmc.rochester.edu/register.html. | ||
| ### License | ||
|
|
||
| ##### Installation of ssHMM: | ||
|
|
||
| 1. Download ssHMM from this page | ||
| 2. Install ssHMM: | ||
|
|
||
| ```bash | ||
| #as root | ||
| sudo python setup.py install | ||
|
|
||
| #as non-root | ||
| python setup.py install --user | ||
| ``` | ||
| This will install ssHMM and the following python package dependencies: numpy, graphviz, pygraphviz, weblogo, forgi. If setuptools fails to install any of the dependencies, try to install it separately (e.g. with `sudo pip install numpy`). | ||
|
|
||
|
|
||
| ### Preprocessing a CLIP-Seq dataset: *preprocess_dataset* | ||
|
|
||
| **usage**: preprocess_dataset [-h] [--genome GENOME] [--min_length MIN_LENGTH] | ||
| [--max_length MAX_LENGTH] | ||
| directory dataset_name jump_to min_score | ||
|
|
||
| **positional arguments**: | ||
| * directory: root directory for data | ||
| * dataset_name: dataset name | ||
| * jump_to: preprocessing step to jump to (as integer): 1 - filter bed, 2 - shuffle bed, 3 - enlongate bed, 4 - fetch sequences, 5 - format FASTA, 6 - calculate RNA shapes, 7 - calculate RNA structures | ||
| * min_score: minimum score for binding site (default: 0.0) | ||
|
|
||
| **optional arguments**: | ||
| * -h, --help: show this help message and exit | ||
| * --genome GENOME: genome version to use (default: hg19) | ||
| * --min_length MIN_LENGTH: minimum binding site length (default: 8) | ||
| * --max_length MAX_LENGTH: maximum binding site length (default: 75) | ||
|
|
||
|
|
||
| This script prepares a CLIP-Seq dataset in BED format for the training of ssHMM. The following preprocessing steps are taken: | ||
| 1 - Filter (positive) BED file | ||
| 2 - Shuffle (positive) BED file to generate negative dataset | ||
| 3 - Enlongate positive and negative BED files for later structure prediction | ||
| 4 - Fetch genomic sequences for elongated BED files | ||
| 5 - Produce FASTA files with genomic sequences in viewpoint format | ||
| 6 - Calculate RNA shapes | ||
| 7 - Calculate RNA structures | ||
|
|
||
| A root directory for the datasets and a dataset name (e.g., the protein name) has to be given. The following files will be created in the root directory and its subdirectories: | ||
| - ``/bed/<dataset_name>/positive_raw.bed`` - positive BED file from CLIP-Seq experiment | ||
| - ``/bed/<dataset_name>/positive.bed`` - filtered positive BED file | ||
| - ``/bed/<dataset_name>/negative.bed`` - filtered negative BED file | ||
| - ``/bed/<dataset_name>/positive_long.bed`` - elongated positive BED file | ||
| - ``/bed/<dataset_name>/negative_long.bed`` - elongated negative BED file | ||
| - ``/temp/<dataset_name>/positive_long.fasta`` - genomic sequences of elongated positive BED file | ||
| - ``/temp/<dataset_name>/negative_long.fasta`` - genomic sequences of elongated negative BED file | ||
| - ``/fasta/<dataset_name>/positive.fasta`` - positive genomic sequences in viewpoint format | ||
| - ``/fasta/<dataset_name>/negative.fasta`` - negative genomic sequences in viewpoint format | ||
| - ``/shapes/<dataset_name>/positive.txt`` - secondary structures of positive genomic sequence (predicted by RNAshapes) | ||
| - ``/shapes/<dataset_name>/negative.txt`` - secondary structures of negative genomic sequence (predicted by RNAshapes) | ||
| - ``/structures/<dataset_name>/positive.txt`` - secondary structures of positive genomic sequence (predicted by RNAstructures) | ||
| - ``/structures/<dataset_name>/negative.txt`` - secondary structures of negative genomic sequence (predicted by RNAstructures) | ||
|
|
||
| The preprocessing step to begin with can be chosen. For each step, the files generated by the previous step need to be present. To execute all steps, only the positive_raw.bed must be present. For the filtering step, the minimum score and binding site lengths can be defined with parameters. | ||
|
|
||
| **IMPORTANT**: To fetch genomic sequences in step 4, the following file must be present in the genomes/ subdirectory: | ||
|
|
||
| - ``/genomes/[version]/[version].genome`` -> BED file defining the size of the chromosomes | ||
| - ``/genomes/[version]/UCSCGenesTrack.bed`` -> BED file defining the gene intervals | ||
| - ``/genomes/[version]/[version].fa`` -> FASTA file containing the human genome | ||
|
|
||
| The version of the genome can be given as an optional parameter. It defaults to 'hg19'. The files for the ``genomes/`` directory can be obtained from UCSC: | ||
|
|
||
| ``/genomes/[version]/[version].genome``: | ||
| -> download from ``http://hgdownload.soe.ucsc.edu/downloads.html#human`` (Full data set), e.g. ``http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.chrom.sizes`` | ||
|
|
||
| ``/genomes/[version]/UCSCGenesTrack.bed``: | ||
| -> download in table browser (http://genome.ucsc.edu/cgi-bin/hgTables); choose most recent GENCODE track (currently GENCODE Gene V24lift37->Basic (for hg19) and All GENCODE V24->Basic (for hg38)) and 'BED' as output format | ||
|
|
||
| ``/genomes/[version]/[version].fa``: | ||
| -> download chromosomes from ``http://hgdownload.soe.ucsc.edu/downloads.html``; e.g. ``wget --timestamping 'ftp://hgdownload.cse.ucsc.edu/goldenPath/hg38/chromosomes/*'``; concatenate chromosomes with cat and print into .fa file (e.g. with ``zcat chr* > hg19.fa``) | ||
|
|
||
| ### Train ssHMM on a CLIP-Seq dataset: *train_seqstructhmm* | ||
|
|
||
| **usage**: train_seqstructhmm [-h] [--motif_length MOTIF_LENGTH] [--baum_welch] | ||
| [--flexibility FLEXIBILITY] | ||
| [--block_size BLOCK_SIZE] [--threshold THRESHOLD] | ||
| [--job_name JOB_NAME] | ||
| [--output_directory OUTPUT_DIRECTORY] | ||
| [--termination_interval TERMINATION_INTERVAL] | ||
| [--write_model_state] [--only_best_shape] | ||
| training_sequences training_structures | ||
|
|
||
| **positional arguments**: | ||
| * training_sequences: FASTA file storing the training sequences | ||
| * training_shapes: FASTA file storing the training RNA shapes | ||
|
|
||
| **optional arguments**: | ||
| * -h, --help: show this help message and exit | ||
| * --motif_length MOTIF_LENGTH, -n MOTIF_LENGTH: length of the motif that shall be found (default: 6) | ||
| * --baum_welch, -b: should the model be initialized with a Baum-Welch optimized sequence motif (default: yes) | ||
| * --flexibility FLEXIBILITY, -f FLEXIBILITY: greedyness of Gibbs sampler: model parameters are sampled from among the top f configurations (default: f=10), set f to 0 in order to include all possible configurations | ||
| * --block_size BLOCK_SIZE, -s BLOCK_SIZE: number of sequences to be held-out in each iteration (default: 1) | ||
| * --threshold THRESHOLD, -t THRESHOLD: the iterative algorithm is terminated if this reduction in sequence structure loglikelihood is not reached for any of the 3 last measurements (default: 10) | ||
| * --job_name JOB_NAME, -j JOB_NAME: name of the job (default: "job") | ||
| * --output_directory OUTPUT_DIRECTORY, -o OUTPUT_DIRECTORY: directory to write output files to (default: current directory) | ||
| * --termination_interval TERMINATION_INTERVAL, -i TERMINATION_INTERVAL: produce output every i iterations (default: i=100) | ||
| * --write_model_state, -w: write model state every i iterations | ||
| * --only_best_shape: train only using best structure for each sequence (default: use all structures) | ||
|
|
||
| This script trains an hidden Markov model for the sequence-structure binding preferences of an RNA-binding protein. The model is trained on sequences and structures from a CLIP-seq experiment given in two FASTA-like files. | ||
| During the training process, statistics about the model are printed on stdout. In every iteration, the current model and a visualization of the model can be stored in the output directory. | ||
| The training process terminates when no significant progress has been made for three iterations. | ||
|
|
||
|
|
||
| ### Train ssHMM on a batch of CLIP-Seq datasets: *batch_seqstructhmm* | ||
|
|
||
| **usage**: batch_seqstructhmm [-h] [--cores CORES] [--motif_length MOTIF_LENGTH] | ||
| [--baum_welch] [--flexibility FLEXIBILITY] | ||
| [--block_size BLOCK_SIZE] [--threshold THRESHOLD] | ||
| [--termination_interval TERMINATION_INTERVAL] | ||
| data_directory proteins batch_directory | ||
|
|
||
| **positional arguments**: | ||
| * data_directory: data directory (must have the following subdirectories: fasta/, shapes/, structures/ | ||
| * proteins: list of RNA-binding proteins to analyze (surrounded by quotation marks, separated by whitespace) | ||
| * batch_directory: directory for batch output | ||
|
|
||
| **optional arguments**: | ||
| * -h, --help: show this help message and exit | ||
| * --cores CORES: number of cores to use (if not given, all cores are used) | ||
| * --motif_length MOTIF_LENGTH, -n MOTIF_LENGTH: length of the motifs that shall be found (default: 6) | ||
| * --baum_welch, -b: should the models be initialized with a Baum-Welch optimized sequence motif (default: yes) | ||
| * --flexibility FLEXIBILITY, -f FLEXIBILITY: greedyness of Gibbs sampler: model parameters are sampled from among the top f configurations (default: f=10), set f to 0 in order to include all possible configurations | ||
| * --block_size BLOCK_SIZE, -s BLOCK_SIZE: number of sequences to be held-out in each iteration (default: 1) | ||
| * --threshold THRESHOLD, -t THRESHOLD: the iterative algorithm is terminated if this reduction in sequence structure loglikelihood is not reached for any of the 3 last measurements (default: 10) | ||
| * --termination_interval TERMINATION_INTERVAL, -i TERMINATION_INTERVAL: produce output every <i> iterations (default: i=100) | ||
|
|
||
| This script trains multiple Hidden Markov models for the sequence-structure binding preferences of a given set of RNA-binding protein. The models are trained on sequences and structures in FASTA format located in a given data directory. | ||
| During the training process, statistics about the models are printed on stdout. In every iteration, the current model and a visualization of the model are stored in the batch directory. | ||
| The training processes terminate when no significant progress has been made for three iterations. | ||
| The project is licensed under the GNU General Public License. |