RNA-Seq
Gene Expression
Challenges
High-throughput RNA-Seq analysis of the cellular transcriptome has become largely standardized over the years. Nonetheless, many options exist to deal with various steps and issues along the way, that can have a dramatic impact on the interpretation and reliability of the results.
- Quality control (Contamination with other organisms, rRNA depletion, mtRNA depletion, removal of sequencing adapter/primer)
- Optimal mapping of spliced reads (multi-mapping, PCR duplicates)
- Optional batch correction (count matrix, contrast)
- Incremental annotation including multiple databases (Ensembl/Gencode, UniProt, Pathways/Ontologies)
- Production of abstract visualizations to assess data quality and depict results (PCA, GSEA, Locus plots, Venn/UpsetR)
- Assembly of tables/figures/methods into concise and user-friendly collections (Excel, Powerpoint)
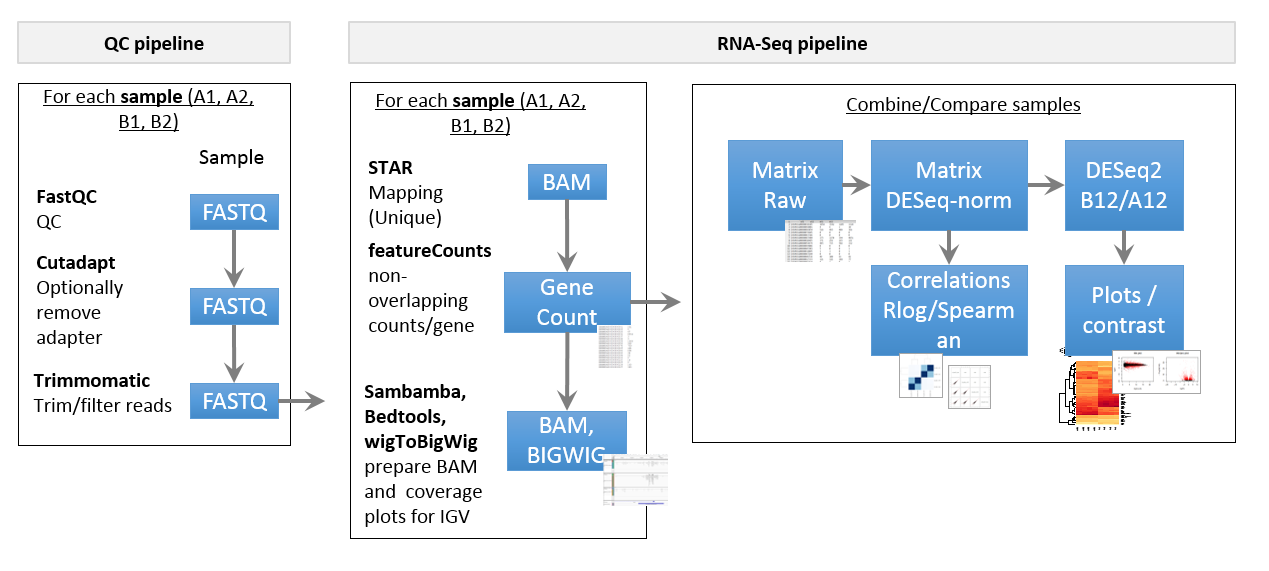
Pipeline
Results
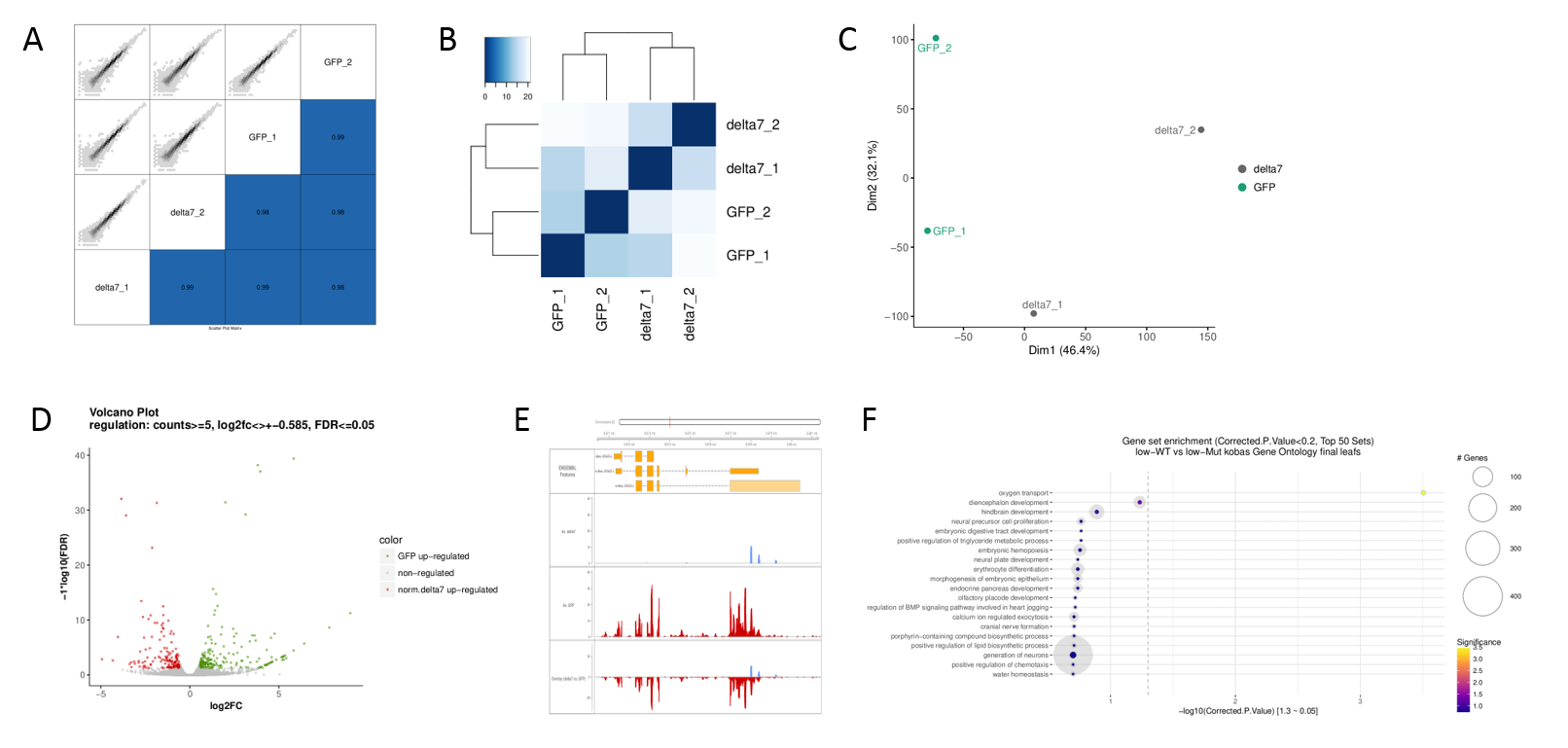 Typical plots produced by the pipeline. A) Scatterplot of all expressed gene counts including spearman correlation, B) Hierarchical clustering of all rlog-transformed gene counts per sample, C) PCA, D) Volcano plot for each contrast, E) Coverage of differentially expressed genes, F) Gene set enrichment analyses.
Typical plots produced by the pipeline. A) Scatterplot of all expressed gene counts including spearman correlation, B) Hierarchical clustering of all rlog-transformed gene counts per sample, C) PCA, D) Volcano plot for each contrast, E) Coverage of differentially expressed genes, F) Gene set enrichment analyses.