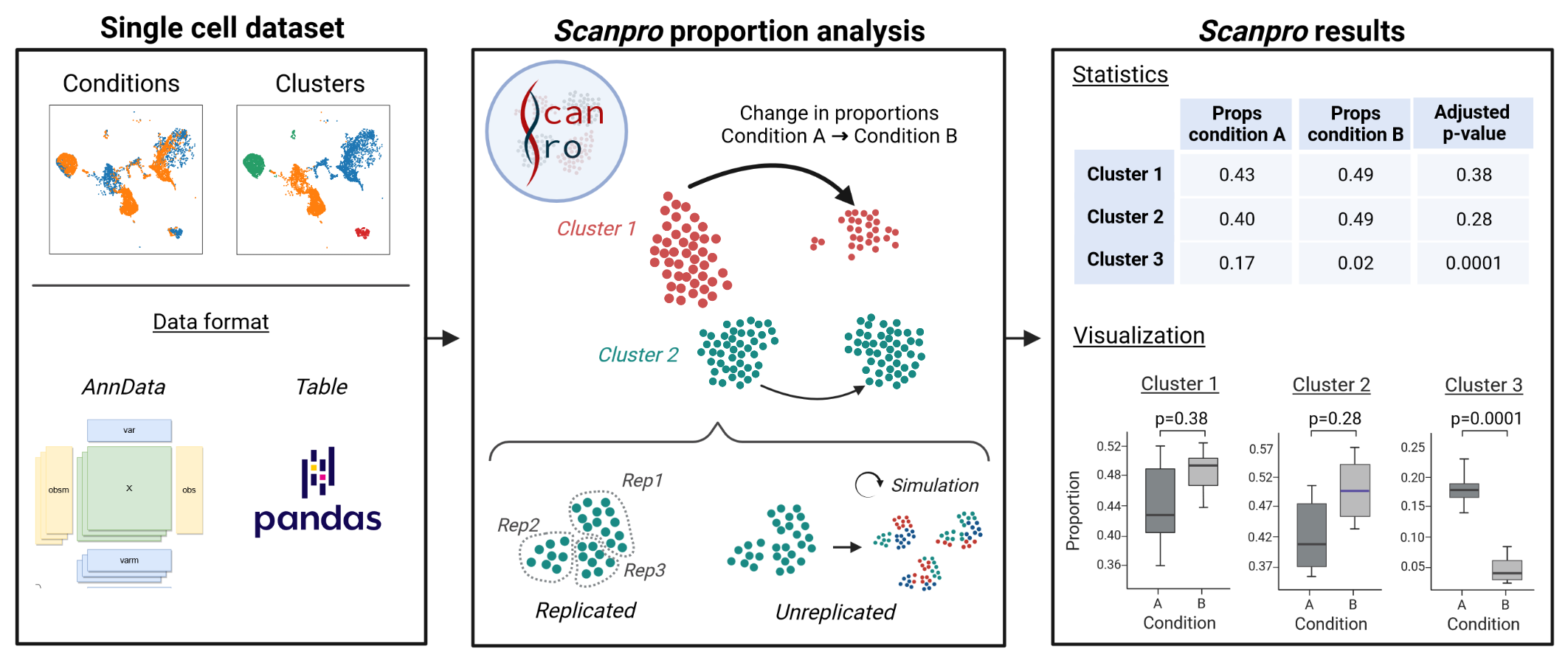
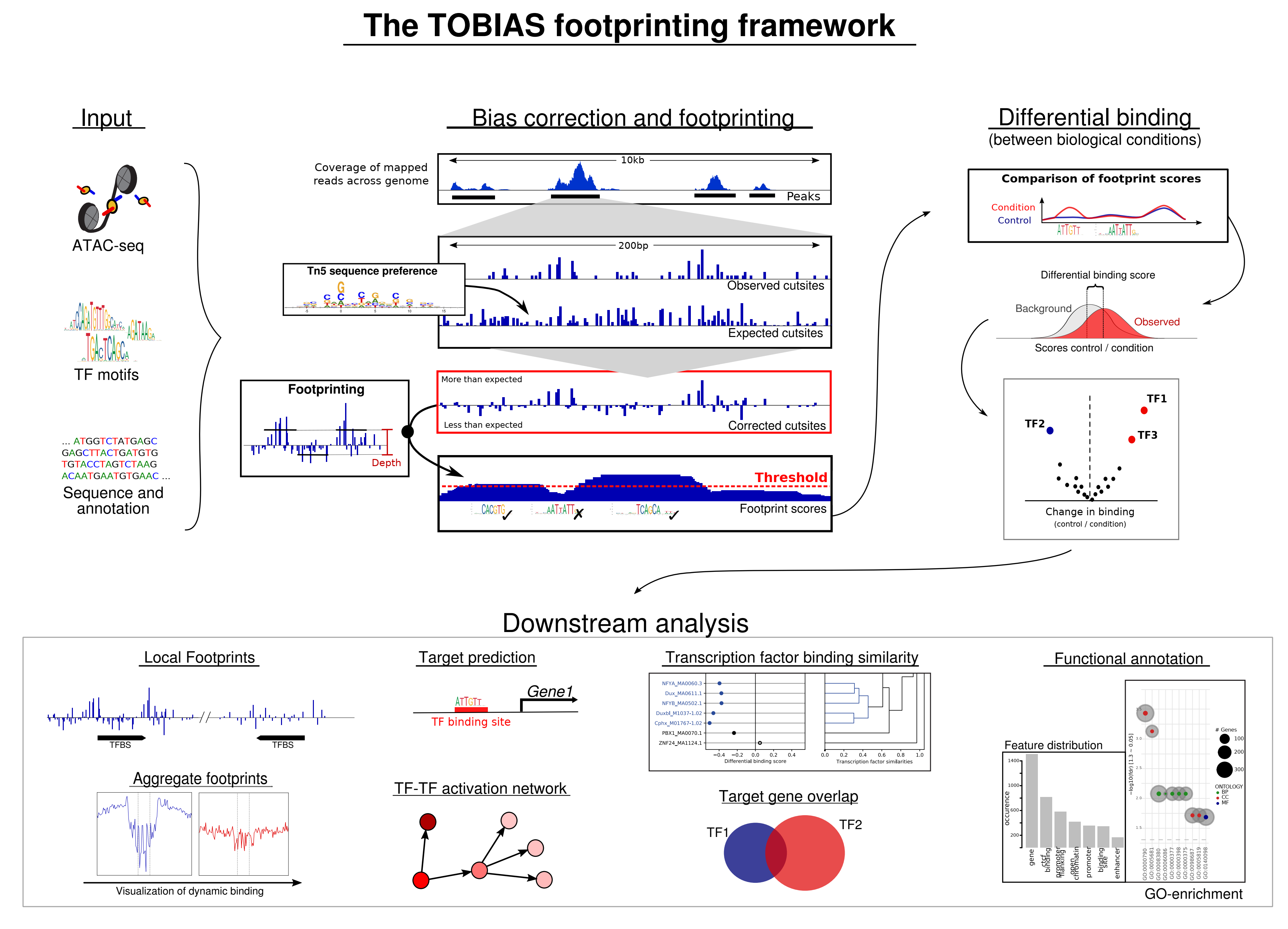
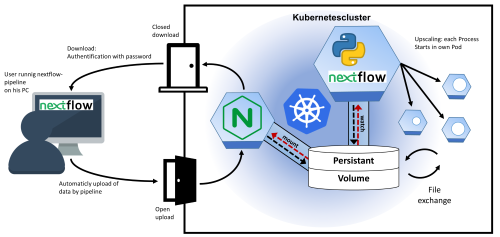
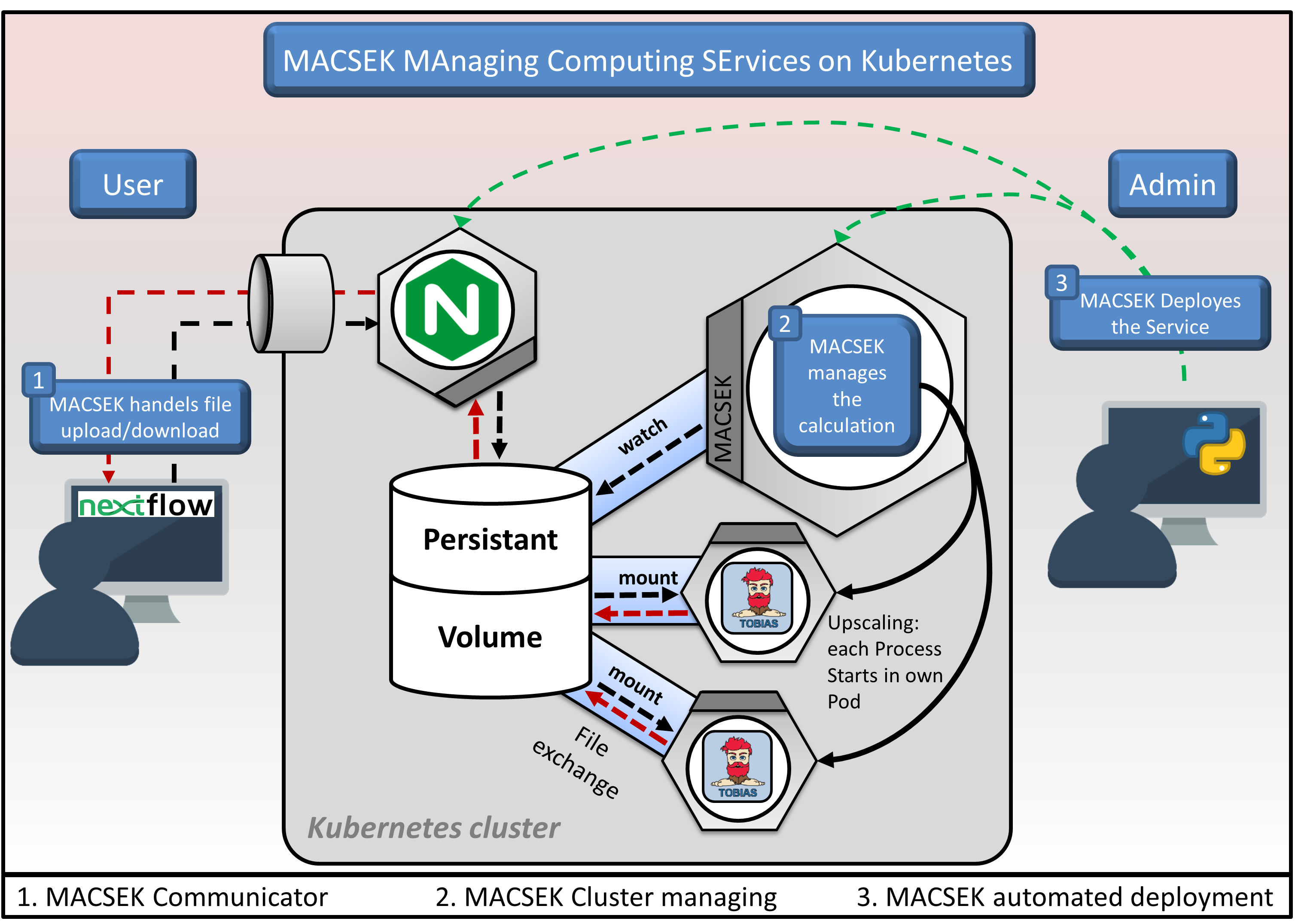
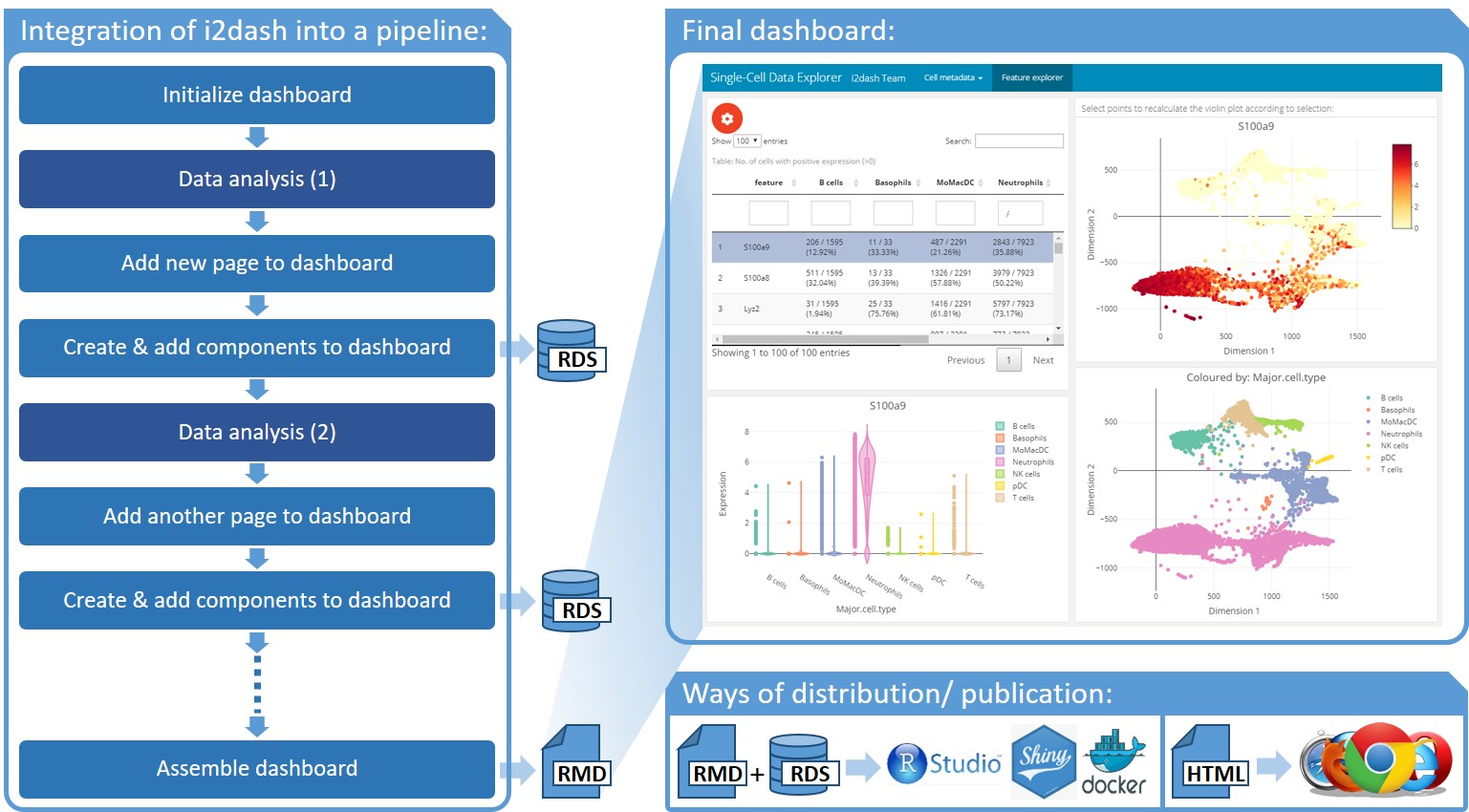
Scanpro is a modular tool for proportion analysis, seamlessly integrating into widely accepted frameworks in the python environment. Scanpro is fast, accurate, support datasets without replicates, and is intended to be used by bioinformatics experts and beginners.
Main Features
Versatility for Replicated and Unreplicated Data
Seamless Integration into Python Ecosystems
Comprehensive Analysis and Visualization
Fast Performance and Robustness
Scanpro can be run on the data using the widely accepted AnnData class object and thus integrated into the Scanpy (scRNAseq), Episcanpy (scATAC), and MUON (multiomics) ecosystems in Python. In addition, a table of cells with annotations in Pandas format is supported. During the analysis, Scanpro uses the number of cells within each condition to estimate whether the cells have different composition in either of the clusters. When the data is replicated, Scanpro applies a Python implementation of the empirical Bayes method presented in the propeller tool. However, when the data is unreplicated, Scanpro offers a robust method to simulate pseudo-replicates by splitting the original samples into multiple replicates using bootstrapping without replacement, which extends the usability of the tool to non-replicated datasets. While this method cannot replicate the biological variance of real replicates, the randomized bootstrapping explores the possibilities that the observed changes in cluster sizes arose by chance. In order to control for outliers of the randomized splitting, the pseudo-replication method is run 100 times and the median p-values for each cluster are calculated. After the analysis, Scanpro reports final statistics, as well as matrices for cell proportions, experimental design, and integrated plotting methods to visualize proportions. These visualizations include a box plot overview of samples (either original or simulated), which can be used to visually confirm differences in cell proportions per cluster. Moreover, Scanpro provides the possibility to restrict the analysis to certain conditions of interest, add covariates per sample as well as support multi-condition comparison using ANOVA. Scanpro is intended to be used at various levels of bioinformatic proficiency by providing exemplary jupyter notebooks and an extended manual within the public code repository